# 生物信息学入门 {#sec-bioinformatics}
```{r}
#| label: check-packages
#| message: false
# 设置CRAN镜像为中国合肥镜像
options(repos = c(CRAN = "https://mirrors.ustc.edu.cn/CRAN/"))
# 检查并安装BiocManager
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
# 设置Bioconductor镜像为中科大镜像
options(BioC_mirror = "https://mirrors.ustc.edu.cn/bioc")
# 检查并安装所需的R包
required_packages <- c(
"tidyverse", # 数据处理和可视化
"pheatmap", # 热图
"igraph" # 网络分析
)
# 检查并安装CRAN包
new_packages <- required_packages[!(required_packages %in% installed.packages()[,"Package"])]
if(length(new_packages)) install.packages(new_packages, dependencies = TRUE)
# 安装Bioconductor包
bioc_packages <- c(
"GEOquery", # GEO数据获取
"limma", # 差异表达分析
"clusterProfiler", # 通路富集分析
"org.Hs.eg.db", # 人类基因注释
"enrichplot" # 富集分析可视化
)
# 安装缺失的Bioconductor包
BiocManager::install(bioc_packages[!(bioc_packages %in% installed.packages()[,"Package"])],
site_repository = "https://mirrors.ustc.edu.cn/bioc",
update = FALSE,
ask = FALSE)
# 加载所需的包
invisible(lapply(c(required_packages, bioc_packages), library, character.only = TRUE))
```
## 基因表达数据分析 {#sec-gene-expression}
### GEO数据库数据获取 {#sec-geo-data}
```{r}
#| label: setup
#| message: false
library(limma)
library(tidyverse)
library(pheatmap)
library(enrichplot)
library(clusterProfiler)
library(org.Hs.eg.db)
# 创建模拟表达数据
set.seed(123)
n_genes <- 1000
n_samples <- 20
# 生成表达矩阵
expr_matrix <- matrix(rnorm(n_genes * n_samples), nrow = n_genes, ncol = n_samples)
rownames(expr_matrix) <- paste0("gene", 1:n_genes)
colnames(expr_matrix) <- paste0("sample", 1:n_samples)
# 创建分组信息
groups <- factor(rep(c("Control", "Treatment"), each = n_samples/2))
# 展示数据维度
dim(expr_matrix)
```
### 差异表达基因分析 {#sec-deg-analysis}
```{r}
#| label: deg-analysis
# 构建设计矩阵
design <- model.matrix(~0 + groups)
colnames(design) <- c("Control", "Treatment")
# 差异分析
fit <- lmFit(expr_matrix, design)
contrast.matrix <- makeContrasts(Treatment - Control, levels = design)
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
# 获取差异基因
deg_results <- topTable(fit2, number = Inf, adjust.method = "BH")
deg_results$gene_id <- rownames(deg_results)
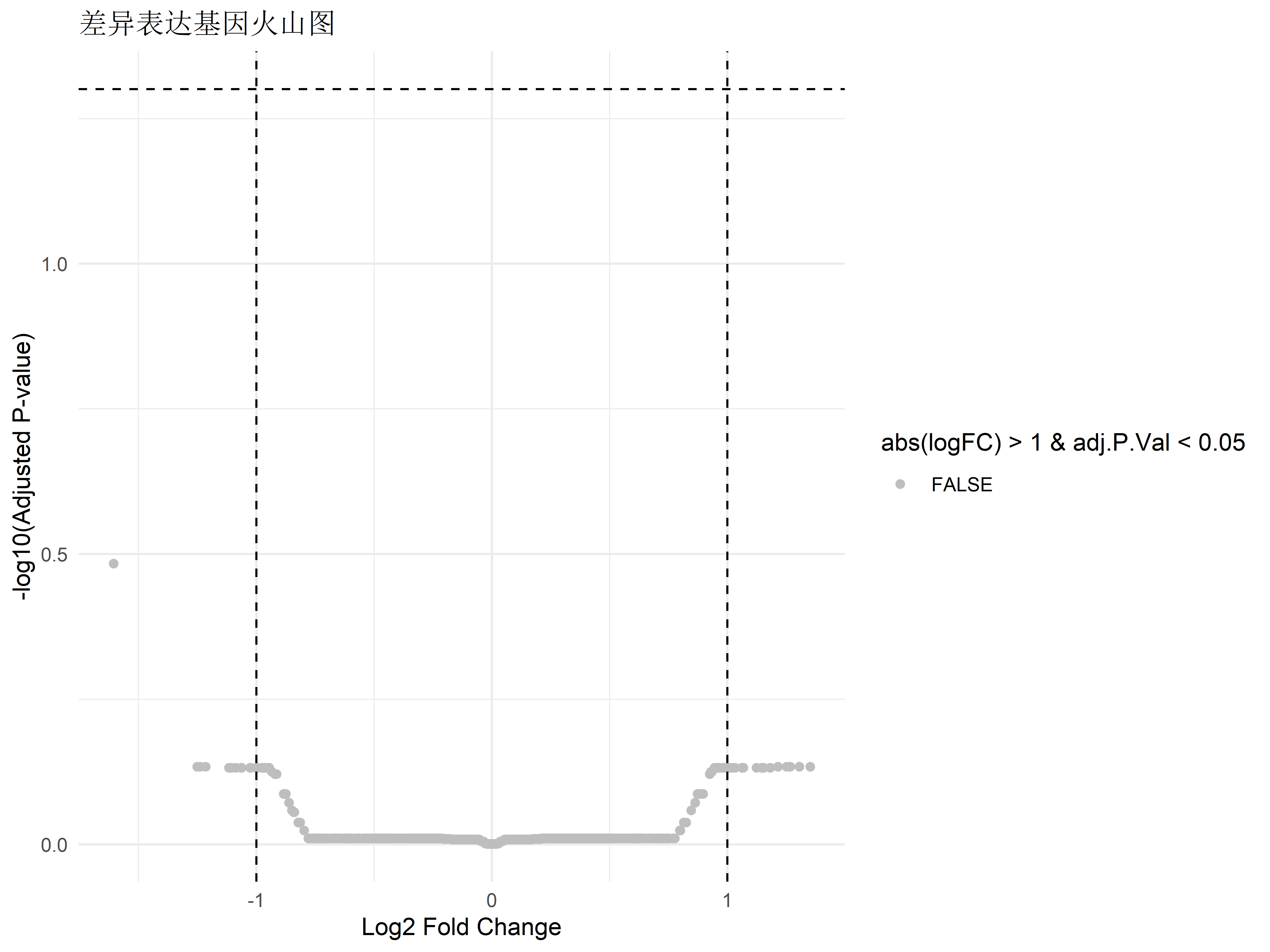
# 可视化火山图
ggplot(deg_results, aes(x = logFC, y = -log10(adj.P.Val))) +
geom_point(aes(color = abs(logFC) > 1 & adj.P.Val < 0.05)) +
geom_vline(xintercept = c(-1, 1), linetype = "dashed") +
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
scale_color_manual(values = c("grey", "red")) +
labs(title = "差异表达基因火山图",
x = "Log2 Fold Change",
y = "-log10(Adjusted P-value)") +
theme_minimal()
```
## 富集分析实践 {#sec-enrichment}
### GO/KEGG分析流程 {#sec-pathway-analysis}
```{r}
#| label: enrichment-analysis
# 创建模拟基因ID(使用ENTREZ ID)
set.seed(123)
all_genes <- sample(keys(org.Hs.eg.db, keytype = "ENTREZID"), n_genes)
rownames(expr_matrix) <- all_genes
# 获取差异基因
deg_genes <- deg_results %>%
filter(abs(logFC) > 1, adj.P.Val < 0.05) %>%
pull(gene_id)
# GO富集分析
ego <- enrichGO(gene = deg_genes,
universe = all_genes,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
# 可视化GO富集结果
if(!is.null(ego) && nrow(ego) > 0) {
dotplot(ego, showCategory = 15) +
ggtitle("GO Biological Process Enrichment")
} else {
message("No significant GO terms found")
}
# 创建热图
if(length(deg_genes) > 0) {
top_genes <- head(deg_genes, 50)
heatmap_data <- expr_matrix[top_genes, ]
# 创建分组注释
annotation_col <- data.frame(
Group = groups,
row.names = colnames(expr_matrix)
)
# 绘制热图
pheatmap(heatmap_data,
scale = "row",
show_rownames = TRUE,
annotation_col = annotation_col,
main = "Top 50 差异表达基因热图")
} else {
message("No significant genes found for heatmap")
}
```
### 结果可视化(气泡图) {#sec-visualization}
```{r}
#| label: pathway-viz
# 如果有显著的富集结果,创建气泡图
if(!is.null(ego) && nrow(ego) > 0) {
# 获取前10个富集通路
top_pathways <- as.data.frame(ego) %>%
slice_head(n = 10)
# 创建富集分析气泡图
ggplot(top_pathways,
aes(x = Count, y = reorder(Description, Count))) +
geom_point(aes(size = Count, color = p.adjust)) +
scale_color_gradient(low = "red", high = "blue") +
labs(title = "GO富集分析结果",
x = "基因数量",
y = "GO Term") +
theme_minimal() +
theme(axis.text.y = element_text(size = 8))
} else {
message("No significant pathways for visualization")
}
# 创建基因表达模式图
if(length(deg_genes) > 0) {
# 选择表达变化最显著的基因
top_varied_genes <- deg_results %>%
arrange(desc(abs(logFC))) %>%
slice_head(n = 20) %>%
pull(gene_id)
# 准备数据
expression_data <- expr_matrix[top_varied_genes, ] %>%
scale() %>% # 对每个基因进行z-score标准化
t() %>% # 转置矩阵
as.data.frame() %>%
mutate(Sample = rownames(.)) %>%
gather(Gene, Expression, -Sample)
# 创建表达模式图
ggplot(expression_data, aes(x = Sample, y = Gene)) +
geom_tile(aes(fill = Expression)) +
scale_fill_gradient2(low = "blue", mid = "white", high = "red") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 45, hjust = 1),
axis.text.y = element_text(size = 8)) +
labs(title = "Top 20 差异表达基因表达模式",
x = "样本",
y = "基因")
} else {
message("No significant genes for visualization")
}
```
## 蛋白质组学基础 {#sec-proteomics}
### 质谱数据预处理 {#sec-ms-preprocessing}
```{r}
#| label: proteomics-demo
#| eval: false
# 注:以下代码仅作演示,需要实际的质谱数据才能运行
library(MSnbase)
library(MALDIquant)
# 读取质谱数据
raw_data <- readMSData("ms_data.mzML", mode = "onDisk")
# 数据预处理流程
processed_data <- raw_data %>%
# 降噪
removeBaseline() %>%
# 峰对齐
alignRt() %>%
# 峰检测
findPeaks() %>%
# 定量
quantify()
```
### 蛋白质互作网络 {#sec-protein-network}
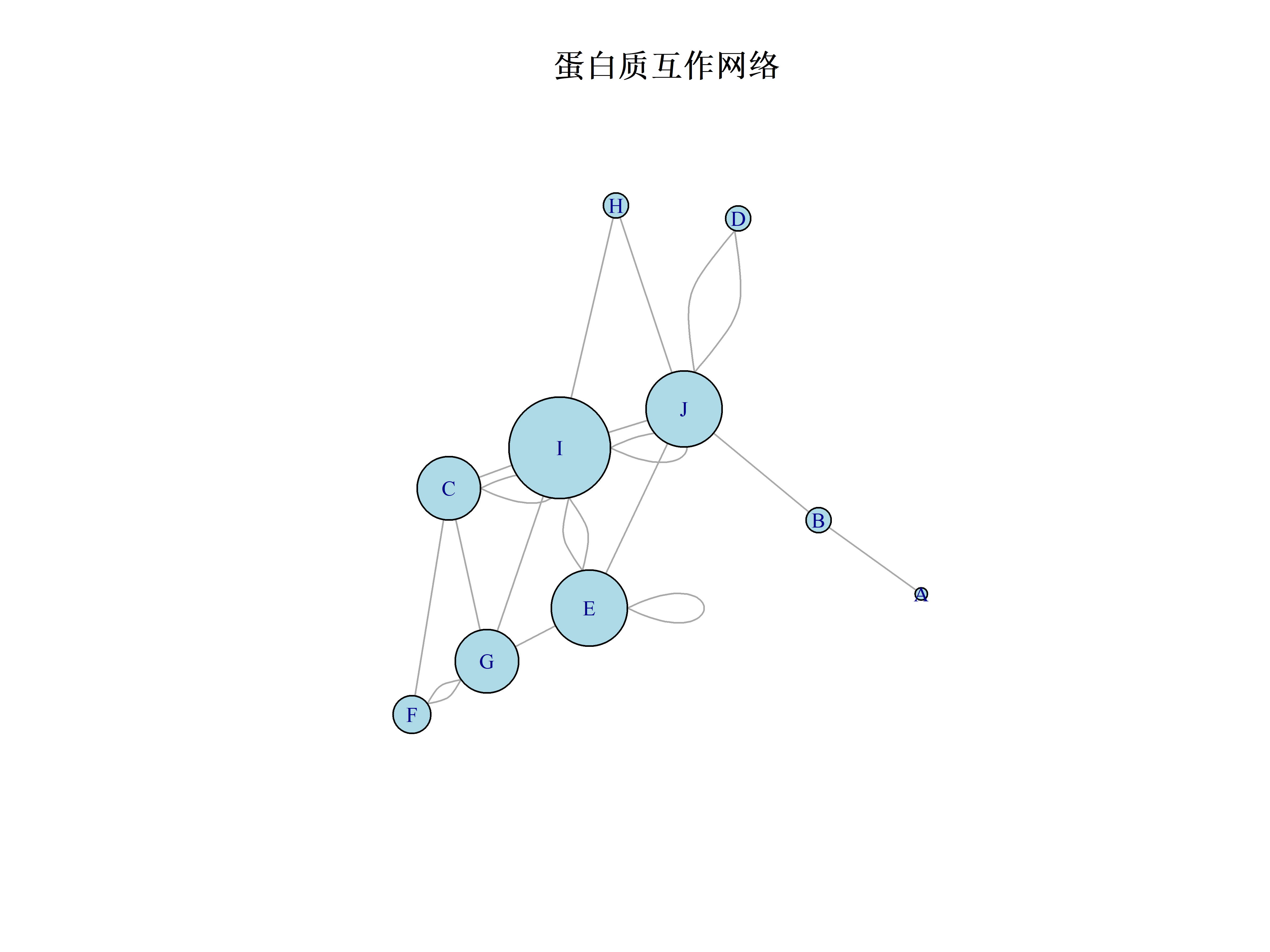
```{r}
#| label: protein-network
library(igraph)
# 创建示例蛋白质互作数据
set.seed(123)
interactions <- data.frame(
protein1 = sample(LETTERS[1:10], 20, replace = TRUE),
protein2 = sample(LETTERS[1:10], 20, replace = TRUE)
)
# 构建网络
network <- graph_from_data_frame(interactions, directed = FALSE)
# 计算网络特征
degree <- degree(network)
betweenness <- betweenness(network)
# 可视化网络
plot(network,
vertex.size = degree * 5,
vertex.label.cex = 0.8,
vertex.color = "lightblue",
main = "蛋白质互作网络")
```
::: callout-tip
## 练习
1. 下载并分析一个GEO数据集
2. 进行差异表达分析和通路富集
3. 创建基因表达热图
4. 构建蛋白质互作网络
:::
## 本章小结
在本章中,我们学习了:
1. 如何获取和处理基因表达数据
2. 差异表达分析的基本流程
3. 通路富集分析方法
4. 蛋白质组学数据分析基础
下一章,我们将通过综合案例来应用所学知识。